지난 세 번의 포스팅을 통해서 몬테 카를로 시뮬레이션에 대해서 알아봤습니다.
https://studyingrabbit.tistory.com/33?category=911605
몬테 카를로 시뮬레이션(Monte Carlo Simulation) 의 이해 : 원주율값 구하기 (+파이썬 시뮬레이션 코드)
몬테카를로 시뮬레이션(혹은 알고리듬)이란 무엇인지를 정의하기란 매우 어려운데, 그 이유는 이 방법론을 수학과 응용수학 분야에서 매우 광범위하게 사용하기 때문입니다. (1 )이 시뮬레이션
studyingrabbit.tistory.com
https://studyingrabbit.tistory.com/34?category=911605
파이썬을 이용한 몬테 카를로 적분(Monte Carlo Integration) 구현
이 포스팅에서는 지난 시간에 설명한 몬테 카를로 시뮬레이션 방법을 프로그램 언어 파이썬으로 구현해 보도록 하겠습니다. 적분은 몬테 카를로 시뮬레이션을 이용해서 할 수 있는 것의 가장
studyingrabbit.tistory.com
https://studyingrabbit.tistory.com/35?category=911605
메트로폴리스-헤이스팅스 알고리듬을 활용한 분포 생성 및 이를 이용한 몬테 카를로 적분
기각 샘플링 기반 몬테 카를로 적분이 부정확한 예시 지난 시간에 소개한 기각 샘플링 방법을 통해서 $$A = \int_{-\infty}^{\infty} x^2 \frac{1}{\sqrt{2 \pi}} e^{-\frac{x^2}{2}}dx$$ 을 해 보도록 하겠습니다..
studyingrabbit.tistory.com
첫 포스팅에서는 몬테 카를로 시뮬레이션의 기본적인 개념을, 두 번째 포스팅에서는 컴퓨터 언어(파이썬)을 이용하여 몬테 카를로 시뮬레이션을 통한 함수의 적분을, 그리고 세 번째 포스팅에서는 몬테 카를로 시뮬레이션을 효율적으로 하기 위한 샘플링을 생성하는 메트로폴리스-헤이스팅스 알고리듬에 대해서 알아 보았습니다.
몬테 카를로 시뮬레이션은 수학 그 자체로도 매우 재미있지만, 이를 응용하여 물리나 공학의 문제를 푸는데 매우 많이 이용 됩니다. 물리가 전공인 저 역시도 몬테 카를로 시뮬레이션을 물리 문제를 풀기 위한 방법 중 하나로 처음 접했는데요, 특히 물리 분야 중에서도 통계역학 분야에서 몬테 카를로 시뮬레이션은 많이 이용 됩니다.
통계역학은 물리의 분과 중 하나인데, 수 많은 입자로 이루어진 시스템의 열역학적 성질을 연구하는 분야 입니다. 말 그대로 수 많은 입자, 예를들면 아보가드로 수인 $10^{23}$ 개 정도의 입자, 가 이루어진 시스템을 연구합니다. $10^{23}$ 개 정도면 사실 무한대 $\infty$로 취급할 수 있고, 따라서 해석적인 방법으로 연구를 할 때는 "시스템의 크기는 무한히 크고, 시스템을 구성하는 입자의 개수 역시 무한히 많다"고 가정을 합니다. 매우 큰 숫자를 다루기 보다는 무한히 많은 숫자를 다루는 것이 편리할 때가 바로 이때 입니다.
그러나, 물리 문제를 해석적으로 다룰 수 있는 지극히 제한적입니다. 학부 수준의 물리에서는 대부분 해석적으로 풀 수 있는 수준의 간단한 문제를 다루지만, 대학원 이상 연구 수준을 다루는 문제에서는 해석적인 풀이가 불가능한 경우가 대부분입니다. 아주 간단한 시스템이라고 할 수 있는 "중력 상호 작용을 하는 3개 입자로 구성된 시스템(삼체문제)" 도 해석적인 풀이가 불가능합니다. 실제 실험과 관련된 문제는 더더욱 해석적인 풀이는 불가능 합니다. 따라서 컴퓨터의 도움을 받아서 시뮬레이션을 할 수 밖에 없습니다. 몬테 카를로 시뮬레이션이 이때 도입됩니다. 지난 포스팅에서 몬테 카를로 시뮬레이션에 대해서 설명할 때, 간단히 말해서 몬테 카를로 시뮬레이션은 매우 고차원 공간에서의 적분을 할 수 있게 해 주는 알고리듬 이라고 하였습니다. 몬테 카를로 시뮬레이션의 바로 이 성질이 통계역학의 문제를 푸는데 적절한 방법론이 될 수 있게 해 줍니다.
통계역학에서 다루는 가장 간단한 모델을 통해 설명해 보도록 하겠습니다. 통계역학에서 계산하는 핵심적인 물리량 중에서는 분배 함수 (Partition function)이 있습니다. $N$개의 입자로 이루어진 고전역학 시스템의 해밀토니안이
$$H(\vec{p}_i, \vec{q_i}) = K + V = \sum_{i=1}^{N} \frac{1}{2m_i} |\vec{p}_i|^2 + V(\vec{q}_i)$$
로 주어졌다고 할 때, 분배 함수 Z 는
$$Z = \frac{1}{N!h^{3N}} \int e^{-\frac{H}{kT}} d^3p_1 d^3p_2 ... d^3p_N d^3q_1 d^3q_2 ... d^3q_N$$
로 주어집니다. 여기서 $h$는 양자역학에서 나오는 플랑크 상수, $k$는 볼츠만 상수, $T$는 시스템의 온도 입니다. 통계역학에 대한 설명이 필요하지만, 이 포스팅을 그것을 설명하는 포스팅이기 때문에 매우 간단히만 설명을 한다면, 만일 우리가 임의의 온도 $T$에서 위 $Z(T)$ 나 $Z(T)$의 $T$에 대한 미분 등을 계산할 수 있다면 우리는 원리적으로 볼 때 시스템의 모든 통계역학적인 성질을 정확히 계산해 낼 수 있습니다. 온도에 따른 시스템의 평균 에너지, 비열 등 다양한 물리량은 모두 $Z(T)$ 함수로 부터 유도 됩니다.
위에서 가정한 것과 같이 해밀토니안이 운동량항과 위치항으로 정확히 분리가 된다면 $e^{-\frac{H}{kT}} = e^{-\frac{K}{kT}} \cdot e^{-\frac{V}{kT}}$ 와 같이 운동량에 대한 함수와 위치에 대한 함수가 분리되고, 운동량에 대한 함수는 가우스적분을 통해 해석적인 적분이 가능합니다. 이 운동량에 대한 적분은 위치 에너지로 기술되는 시스템에 구체적인 정보와는 상관이 없이 주어지는 값이기 때문에 별다른 문제가 되지 않습니다.
위 분배 함수 적분에서 문제가 되는 것은 위치에 대한 적분입니다. 위치에 대한 적분을 따고 분리해서 다시 쓰면 (비례상수를 제거하고 쓰겠습니다)
$$Z_Q =\int exp\Big(-\frac{V(\vec{p}_1, \vec{p}_2, ..., \vec{p}_N)}{kT}\Big) d^3q_1 d^3q_2 ... d^3q_N $$
가 됩니다. 여기서 $V(\vec{p}_1, \vec{p}_2, ..., \vec{p}_N)$가 매우 복잡한 함수라면 이 식은 해석적으로 적분이 불가능해 집니다. 앞에서 언급한 것 처럼, 교과서 수준의 매우 극단적으로 쉬운 경우가 아니라면 이 식을 해석적으로 적분하는 것은 불가능 합니다. 만일, 단순하지 않은 위치 함수 $V(\vec{p}_1, \vec{p}_2, ..., \vec{p}_N)$에 대해서 위 식을 해석적으로 적분이 가능한 경우를 찾아서 논문을 쓴다면, 한 순간에 한국에서 가장 훌륭한 통계물리학자 될 수 있습니다.
위 적분은 $3N$차원 적분입니다. 입자의 개수 $N$이 10개만 되더라도 30차원 적분이 됩니다. 100개라고 하면 300차원이 됩니다. 따라서 구분구적법 혹은 구분구적법을 효율적으로 발전시킨 방법을 쓴다고 하더라도 수치 적분은 불가능하니다. 지난 포스팅에서도 설명을 하였는데, 각 좌표축 당 $10$개의 포인트의 정도의 매우 성근(coarse) 그리드(grid)를 생각한다 하더라도 300차원 공간의 총 포인트의 개수는 $10^{300}$개가 됩니다. 현재 컴퓨터 기술로는 이 계산을 할 수 없습니다. 따라서 위 $Z_Q$의 계산(적분)은 몬테 카를로 시뮬레이션을 통해 합니다. 좀 더 정확히는 메트로폴리스-헤이스팅스 알고리듬을 이용하여 샘플링을 하고, 샘플링된 입자들의 위치값을 사용하여 원하는 통계물리의 물리량을 계산합니다.
지난 포스팅에서는 1차원 변수 $x$에 대해서 확률 밀도 함수 $p(x) = \frac{1}{\sqrt{2 \pi}}e^{-\frac{x^2}{2}}$ 를 만족하는 $x$의 샘플링 포인트를 메트로폴리스-헤이스팅스 알고리듬을 이용하여 구해봤습니다. 이번 포스팅에서 해야하는 것도 똑 같습니다. 단지 조금 더 복잡해졌을 뿐인데, 변수 $x = (\vec{p}_1, \vec{p}_2, ..., \vec{p}_N)$의 차원은 $3N$ 차원이 되었고, 변수 $x$가 따라야 하는 확률 밀도 함수는 (비례상수를 제외하고 쓰면) $exp\Big(-\frac{V(\vec{p}_1, \vec{p}_2, ..., \vec{p}_N)}{kT}\Big)$ 입니다. 자연상수의 지수가 $-\frac{x^2}{2}$에서 $-\frac{V(\vec{p}_1, \vec{p}_2, ..., \vec{p}_N)}{kT}$로 바뀐 것 빼고는 아무런 차이가 없습니다. 물론 이 차이가 계산 시간을 더 오래 걸리게 하는 것이긴 하지만, 개념적으로는 어려운 부분은 하나도 없다고 해도 됩니다.
구체적인 위치 에너지에 대한 문제를 풀어 보자
간단하면서도 의미 있는 실제 문제를 풀어 보도록 하겠습니다. 1차원 공간에서 $N$개의 입자가 조화 포텐셜(harmonic potential) 에 갖혀 있는 시스템을 생각하겠습니다. 즉, 위치 에너지 항에서 외부 필드에 의한 포텐셜이
$$V_{ext}(x) = \frac{1}{2}m\omega^2 x^2 $$
으로 주어지는 것 입니다. $\omega$값에 이해서 위치 에너지의 크기가 조절됩니다. 입자간 상호작용에 의한 위치에너지는 전기적 위치 에너지, 즉 쿨롱 포텐셜을 가정하도록 하겠습니다. 인력을 가정하면 입자들이 한 곳으로 응축하는 결과가 얻어지기 때문에, 전하가 같아서 서로 밀어내는 힘을 가한다고 하면
$$V_{int}(x_1, x_2) = \frac{k}{|x_1 - x_2|}$$
가 됩니다. 여기서 $k$는 비례상수 입니다. 따라서 $N$개의 입자에 대한 총 위치 에너지를 하나의 식으로 쓰면,
$$V(x_1, x_2, ..., x_N) = \sum_{i=1}^{N} \frac{1}{2}m\omega^2 x^2 + \sum_{\langle i, j \rangle} \frac{k}{|x_i - x_j|}$$
가 됩니다.
우선, 간단한 경우에 대한 계산을 해 보도록 하겠습니다. 상호작용에 대한 위치 에너지가 없는 경우를 먼저 생각하겠습니다. 즉 $V_{int} = 0$ 인 경우 입니다. $k=0$인 경우라고 생각해도 좋습니다. 지난 포스팅에서 다룬 메트로폴리스-헤이스팅스 알고리듬을 이용하여 계산하면 됩니다. 지난 포스팅에서 자유도는 실수값을 가지는 변수 하나였습니다. 이번 문제에서 자유도는 $N$개의 실수 $(x_1, x_2, ..., x_N)$이 됩니다. 편의상 각 실수 $x_i$가 가질 수 있는 값의 범위를 $[x_{min}, x_{max}]$로 제한 하도록 하겠습니다. 이 $(x_1, x_2, ..., x_N)$에 대한 확률 $p(x_1, x_2, ..., x_N)$은 $p(x_1, x_2, ..., x_N) = exp\Big( -\frac{V_{exp}(x_1, x_2, ..., x_N)}{T}\Big)$ 와 같습니다. 앞으로는 편의상 $\vec{x} =(x_1, x_2, ..., x_N)$로 쓰도록 하겠습니다. $p(\vec{x}) = exp\Big( -\frac{V_{exp}(\vec{x})}{T}\Big)$가 되는 것은 통계역학의 볼츠만 분포인데, 왜 이 분포가 얻어지는지에 대해서는 설명을 생략 하도록 하겠습니다.
이제 이 $p(\vec{x})$에 대해서 메트로폴리스-헤이스팅스 알고리듬을 적용하면 됩니다. $\vec{x}$와 $\vec{x}'$을 생성합니다. 그리고 이에 대응 되는 확률 $p(\vec{x})$, $p(\vec{x'})$ 를 계산합니다. $\vec{x} \rightarrow \vec{x}'$ 로 상태를 이동시킬지 말지는 $p(\vec{x}') / p(\vec{x})$ 값에 의해 결정 됩니다.
실제 파이썬 프로그래밍을 통해 위 계산을 구현하였습니다. 계산에 이용된 구체적인 물리 상수 값은 아래와 같습니다.
(1) $N=500$개의 입자가 1차원 공간 $x \in [x_{min} = -10, x_{max} = 10]$에 있다.
(2) 입자들은 외력에 의한 위치 에너지 $V_{ext}(x) = \frac{1}{2}m\omega^2 x^2$ 를 받는다.
(3) 볼츠만 상수 $k_B = 1$, 입자의 질량 $m = 1$, 시스템의 온도 $T = 1$로 간단히 하였다.
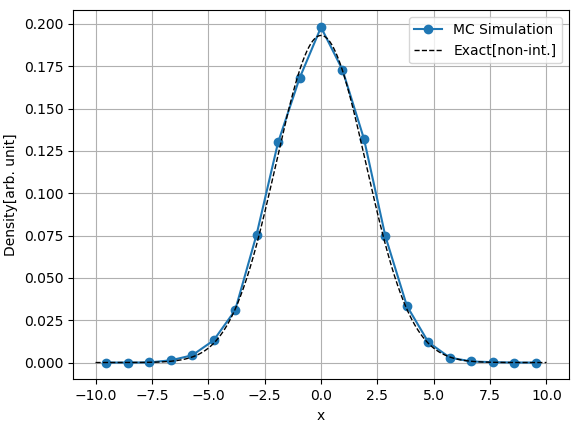
위 조건하에서 입자의 밀도 $n(x)$는 위 그래프의 파란색 실선과 같습니다. 입자들은 서로 상호작용 하지 않기 때문에, 서로 독립이며, 따라서 1개의 입자에 대한 결과에 곱하기 $N = 500$을 한 것과 같은 결과를 줍니다. 상호작용이 없는 경우에는 해석적인 방법으로 볼츠만 팩터를 구할 수 있고, 그 값은 위 그래프에서 점선으로 표시하였습니다. 위 결과를 보면 해석적인 값과 몬테 카를로 시뮬레이션을 통해서 얻은 값이 일치함을 볼 수 있습니다. 열평형 상태에서는 총 에너지가 낮은 곳에 입자의 밀도가 크기 때문에 위치 에너지가 낮은 원점 근처에 많은 입자들이 모여 있음을 볼 수 있습니다. 원점에서 멀어질수록 멀어진 거리의 제곱에 비례해서 총 에너지가 커지기 때문에, $x_{min}$ 혹은 $x_{max}$ 주변의 밀도는 거의 $0$이 됩니다. 사실상 $|x| \gt 7.5$ 에서는 밀도가 $0$입니다.
입자들이 상호작용을 한다면 열평형 상태에서 밀도는 어떻게 바뀔까?
입자와 입자의 상호 작용 위치 에너지 $V_{int}(x_1, x_2) = \frac{k}{|x_1 - x_2|}$를 추가해 보도록 하겠습니다. $k$값의 크기에 따라서 상호 작용의 크기가 달라집니다.
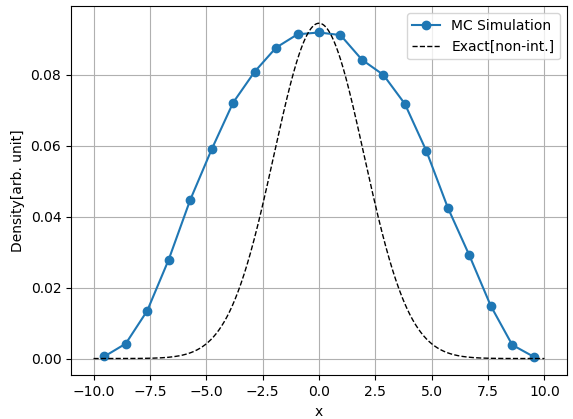
$k = 0.01$ 인 경우의 열평형 상태에서의 입자의 밀도 $n(x)$ 그래프는 위와 같습니다. 입자간 상호작용이 없는 경우에 비해서 원점 근처의 집중도는 조금 떨어졌습니다. 그리고 위와는 달리 $|x| \gt 7.5$ 에서 또한 $x_{min}, x_{max}$ 근처에서도 밀도가 0이 아니라 큰 값을 가집니다. 위치 에너지에 의해서 입자들은 원점 근처로 모이게 되는데, 원점 근처에 많이 모여 있는 입자들은 입자간 상호작용에 의한 위치에너지를 크게 받습니다. 그래서 입자들은 밀도가 낮은 원점에서 멀어지는 곳으로 가려고 하는 "성향"이 생기고, 원점에서 먼 지역에도 입자가 존재하게 돼 밀도가 $0$이 아닌 값을 갖게 됩니다.
위 시스템의 $n(x)$ 값을 이용하여 위치 $x$에 따른 유효 위치에너지(effective potential)을 정의 할 수 있습니다. 이 시스템의 유효 위치에너지는 이 시스템의 입자의 밀도 $n(x)$와 동일한 밀도를 갖게 하는 외부 힘에 대한 위치에너지 라고 정의 할 수 있습니다. 다시 설명하면, 외부 힘에 의한 위치에너지와 입자간 상호작용에 의한 위치에너지가 모두 있는 시스템을 $A$라고 할 때, 이 시스템의 열평형 상태에서의 입자의 밀도를 $n_A(x)$라고 하겠습니다. 또 다른 시스템은 외부 힘에 의한 위치에너지 $v_{eff}(x)$밖에 없는데, 즉 입자간 상호작용을 하지 않을 때, 이 시스템의 입자의 밀도를 $n_B(x)$라 하겠습니다. 이 때, $n_A(x) = n_B(x)$가 되게 하는 $v_{eff}(x)$를 시스템 $A$의 유효 포텐셜이라고 합니다.
정의에 의해서 $v_{eff}(x) = - log(n(x)/T)$가 되고, $n(x)$를 위 그래프의 결과를 이용하면 아래와 같은 $v_{eff}(x)$를 얻게 됩니다.

$|x| \gt 5$ 영역에서, 파란색으로 표시한 유효 위치에너지 값이 외부 힘에 대한 위치 에너지보다 작아짐을 볼 수 있습니다! 약간 직관과는 거리가 있는 결과인데, 유효 위치에너지는 외부 힘에 대한 위치 에너지와 입자간 상호작용에 의한 위치에너지의 합인데, 상호작용에 의한 위치에너지는 척력이라서 양의 값을 갖게 되고, 따라서 적당히 평균을 한다고 하더라도 상호작용에 의한 위치에너지는 양수 값을 갖게 됩니다. 따라서 유효 위치에너지는 외력에 의한 위치에너지보다 커야 할 것 같은데, 실제로는 그렇지 않습니다. $| x | \approx 10$ 근처에서는 거의 절반 밖에 되지 않습니다.
이는 유효 위치에너지가 실제 "위치에너지의 평균" 값으로 정해진 것이 아니라 "동일한 밀도를 주는 위치에너지"로 정의 되었기 때문입니다. 따라서 각 위치에서의 밀도이 차이 값이 유효 위치에너지를 결정하게 되고, 입자간 상호작용이 있는 경우에는 위에서 본 것과 같이 밀도 $n(x)$가 입자간 상호작용이 없는 시스템에 비해서 상대적으로 균일하기 때문에, 위치간 유효 위치에너지 값의 차이가 덜 한것 입니다.
유효 위치에너지는 입자간 상호작용이 있는 시스템을 입자간 상호작용이 없는 시스템인것 처럼 다룰때 도입되는 값입니다. 계산을 하기 위해서나 혹은 개념적으로나 매우 편리한 값인데, 앞에서 언급한 바와 같이 입자간 상호작용이 있는 시스템에서는 $n(x)$를 해석적으로 구하는 것이 불가능하기 때문에, 우리가 한 것과 같이 몬테 카를로 시뮬레이션을 통해서 그 값을 구했습니다.


